この投稿は,iGEM・Synthetic biology(合成生物学)・Japan Advent Calendar 2022の25日目です.
はじめに
メリークリスマス,私,tax_freeはいろいろあって沖縄に来てます.
今日は高校生の時によく行ってたトンカツを食べに行きました.なつかしいね.
今日でiGEM・Synthetic biology(合成生物学)・Japan Advent Calendar 2022も最後の投稿になります.
24日の投稿のクオリティが高かったのでプレッシャーが…
突然ですが,モデリングしてますか?
2022のTokyoTechは,Dryだけでデング熱の血清型ごとの感染者割合を予測するシステムを作っていたので,Wetの系に関するモデリングはしていませんでした. が,他のiGEMチームを見てみると,みんなWetと協力して生成物の構造予測や量の予測をしています.
そして,モデリングはGold要件です.
現状のモデリングについて
十分な数を見ているわけではないですが,ほとんどのチームが一般のODEソルバーを使うか,特別なライブラリを使うのではなく普通にPythonとかを使っていると思います. 数は少ないですが,AlphaFoldを使ってるチームもありました.
これらで実装することの問題点として,Dryの人しか実装できなかったり,他のメンバーと結果の共有がしずらいということがあります.
この記事では,これらの課題を少しでも解決するために,Python向けのモデリング補助ライブラリについて解説説明します.
使うライブラリと具体的な問題例
実行環境は,Colaboratoryです.ソースコードはこれ.
今回使うライブラリはBioCRNpyler(CRNはChemicalReactionNetworkの略)で,SBMLシミュレータにBioscrapeを使います. 参考にしたページは下にまとめておきます.
BioCRNpylerの構造は次のようになっています.
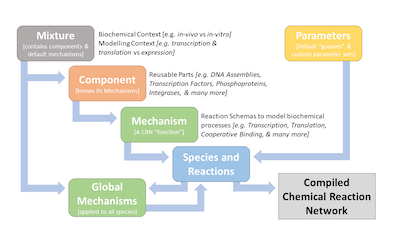
なので,モデルを実装するときは,次に手順に従って実装することになります.
- Mixtureに追加されるComponentとMechanismをインスタンス化します.BioCRNpylerには,酵素や触媒作用,遺伝子制御ネットワークなどのComponentやMechanismがライブラリとして用意されています.なお、Componentは,カスタムしたMechanismsを持つことも,MixtureのMechanismsを利用することも可能です.
- Mixtureを作成しましょう.ComponentやMechanismがあらかじめ組み込まれた様々なMixtureがありますし,空のMixtureを使用することもできます.
- パラメータは,dictかパラメータファイルでMixture(もしくは個別のComponent)に追加することができます.パラメータの初期化をすることで,非常に少ないパラメータでモデルをコンパイルすることができ,後から精度を上げるために微調整することができます.
- CRNにMixtureをコンパイルする.
- コンパイルしたCRNはSBMLとして保存,プロット,シミュレートすることができます.
次のセクションで簡単なサンプルを実装します.
実装方法とできること
今回の記事では,簡単なサンプルとして下の図のような反応のモデリングを考えます.
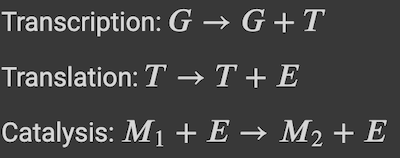
これをBioCRNpylerで実装します.必要なライブラリは,既に定義しているとします.ソースコードを参考にしてください.
まずは,それぞれの文字を定義します.
#M1とM2は代謝物を表すSpeciesである
M1 = Species("M1")
M2 = Species("M2")
G = Species("G")
T = Species("T")
E = Species("E")
次にそれぞれの反応の速度定数を設定します.
#質量作用の法則が働くReactionで反応速度定数は1.0
r_tx = Reaction.from_massaction([G], [G, T], k_forward = 1.0)
r_tl = Reaction.from_massaction([T], [T, E], k_forward = 1.0)
r_cat = Reaction.from_massaction([M1, E], [M2, E], k_forward = 1.0)
最後にこれらの反応を繋げます.
#CRNに保存
CRN = ChemicalReactionNetwork(species = [M1, M2, G, T, E], reactions = [r_tx, r_tl, r_cat])
次に,このCRNによりComponentとMechanismを加えます.
#ComponentとSpeciesを作る
#酵素を作る
E = Enzyme(enzyme = 'E', substrates = [M1], products = [M2])
#DNAアセンブリは転写-翻訳のユニット
G = DNAassembly('G', protein = E.get_species(), promoter = 'P', rbs = 'RBS')
#いくつかのMechanismを追加する
catalysis = BasicCatalysis()
transcription = SimpleTranscription()
translation = SimpleTranslation()
#パラメータはdictで定義できて,ここではデフォルトの値を使います
parameters = {
'kcat' : 1.0,
'ktx' : 1.0,
'ktl' : 1.0
}
#初期状態の作成(TはGによるのでGとM1だけ与える)
initial_concentrations = {
G.dna : 1.0,
M1 : 10.0
}
そして,これらを使ってMixtureを作成します.
#Mixtureを作成する
M = Mixture(mechanisms = [catalysis, transcription, translation], components = [G, E], parameters = parameters)
さて,Mixtureを作成したので,コンパイルしてみましょう.
CRN_compiled = M.compile_crn(initial_concentration_dict = initial_concentrations)
作成したCRN_compiledの時間発展を,Bioscrapeを使ってプロットしてみましょう.横軸を時間,縦軸を濃度として考えます.
#Bioscrapeを使ったCRNのシミュレーションとプロット
timepoints = np.arange(0, 10, 0.1)
results = CRN_compiled.simulate_with_bioscrape_via_sbml(timepoints)
plt.subplot(1, 2, 1)
plt.plot(timepoints, results[str(G.dna)], label = str(G.dna))
plt.plot(timepoints, results[str(G.transcript)], label = str(G.transcript))
plt.plot(timepoints, results[str(G.protein)], label = str(G.protein))
plt.legend()
plt.subplot(1, 2, 2)
plt.plot(timepoints, results[str(M1)], label = str(M1))
plt.plot(timepoints, results[str(M2)], label = str(M2))
plt.legend()
こういう結果が出ると思います.
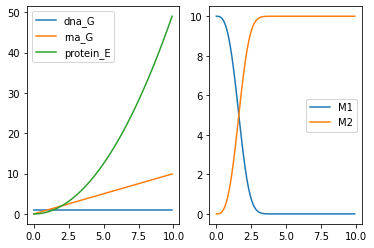
結果について軽く考察すると,最初に示した反応経路から,Gの濃度は一定,Tの濃度は線型,Eの濃度はTの濃度に線型なので放物線になります. なので,図の左側は合ってそうだな,と分かります.また,右側についてもM_1とM_2が対象で,感覚的に指数関数っぽい立ち上がりになりそうです. なので,左も右も合ってそうですね.
最後に,このCRNをグラフにして可視化してみましょう.グラフの表示にはBokehを使います.
from bokeh.plotting import figure, show, output_notebook
plot = render_network_bokeh(CRN,
use_pretty_print = True,
pp_show_rates = False,
pp_show_attributes = False,
pp_show_material = True,
)
output_notebook()
show(plot)
こんな図が表示されると思います.ノードがそれぞれのSpeciesに対応して,エッジの向きが反応の向きに対応しています.
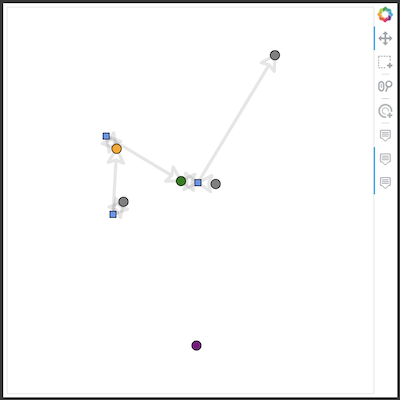
下の方にあるオブジェクトは消す方法が分からんかった…公式サンプルにも載ってるので気にしない,気にしない.
これから
今回は非常に簡単なモデルだったので,BioCRNpylerがラップしてくれる嬉しさ,グラフにしてくれる嬉しさが十分に感じれないかもしれないですが,より複雑なモデルを扱う時には,きっと役にたつはずです.
参考にしたサンプルには,この後にMichaelis–Menten equationに従うようなモデルを考えていましたが,コピペしてもエラーが出てくるので今回の記事には間に合いませんでした.Michaelis–Menten equationの次は,パラメータのAutoReduceをやってくれるサンプルなので,BioCRNpylerはモデルの最適化の方にも使えそうです.次の記事では,これを触ってみようかなぁ…
おわりに
この記事を最後まで読んでくれた方,そしてiGEM・Synthetic biology(合成生物学)・Japan Advent Calendar 2022を読んで書いてくれたiGEMerの方には,とても感謝しています.私がアドベントカレンダーをやりたいと言った時,正直ぜんぜん埋まらないんじゃないかと不安でした.でも,やってみると全部の枠が埋まった(1枠空いたけど)ので,とってもとっても嬉しいです.
最後に,これからもiGEM Japan Community,iGEM TokyoTechのメンバーとして精進するので,応援よろしくおねがいします!
それでは,楽しいクリスマス,年末年始をお過ごしくださいな.